
Uploading data_analysis correctly
Showing
This source diff could not be displayed because it is too large. You can view the blob instead.
{kind=link}
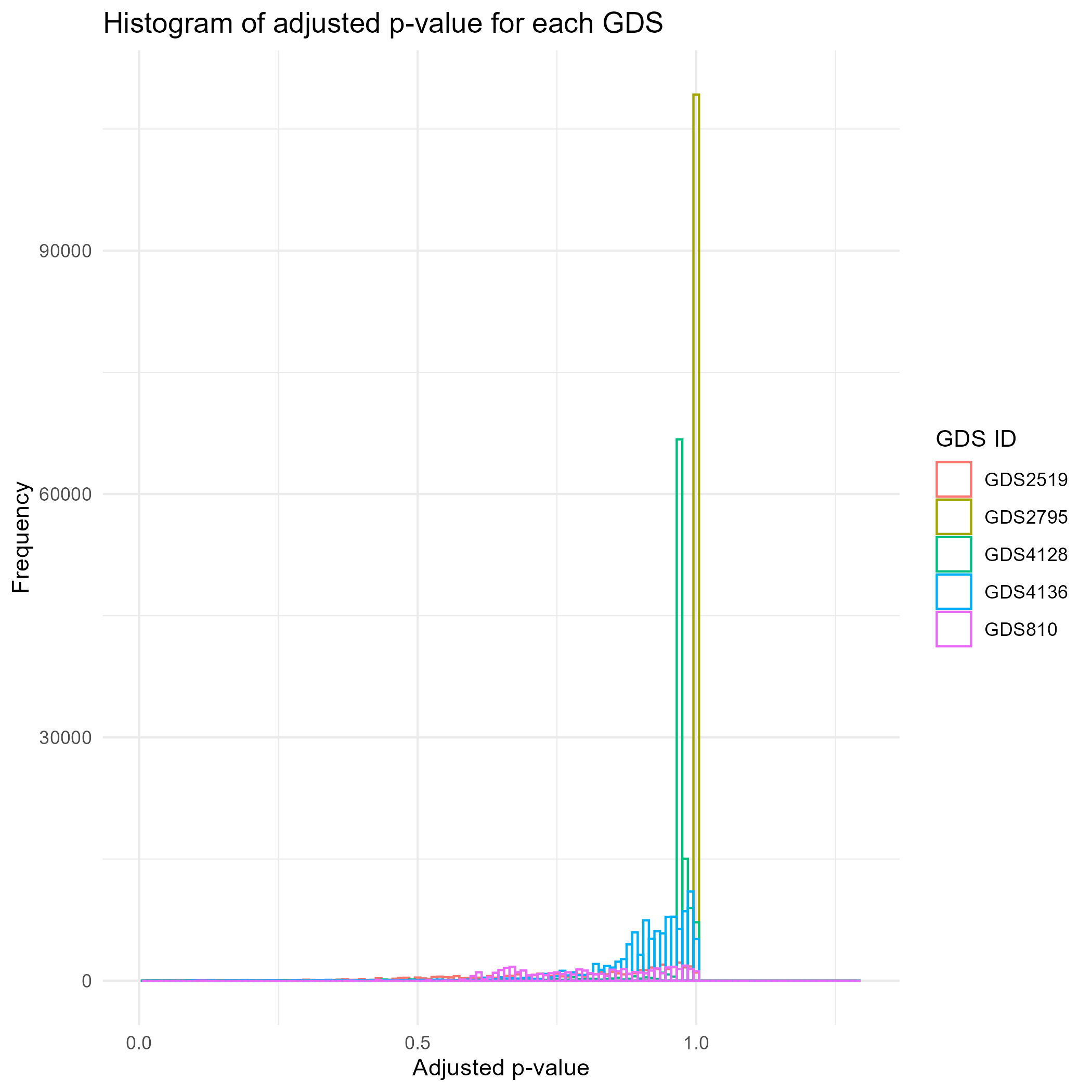
90.4 KB
{kind=link}
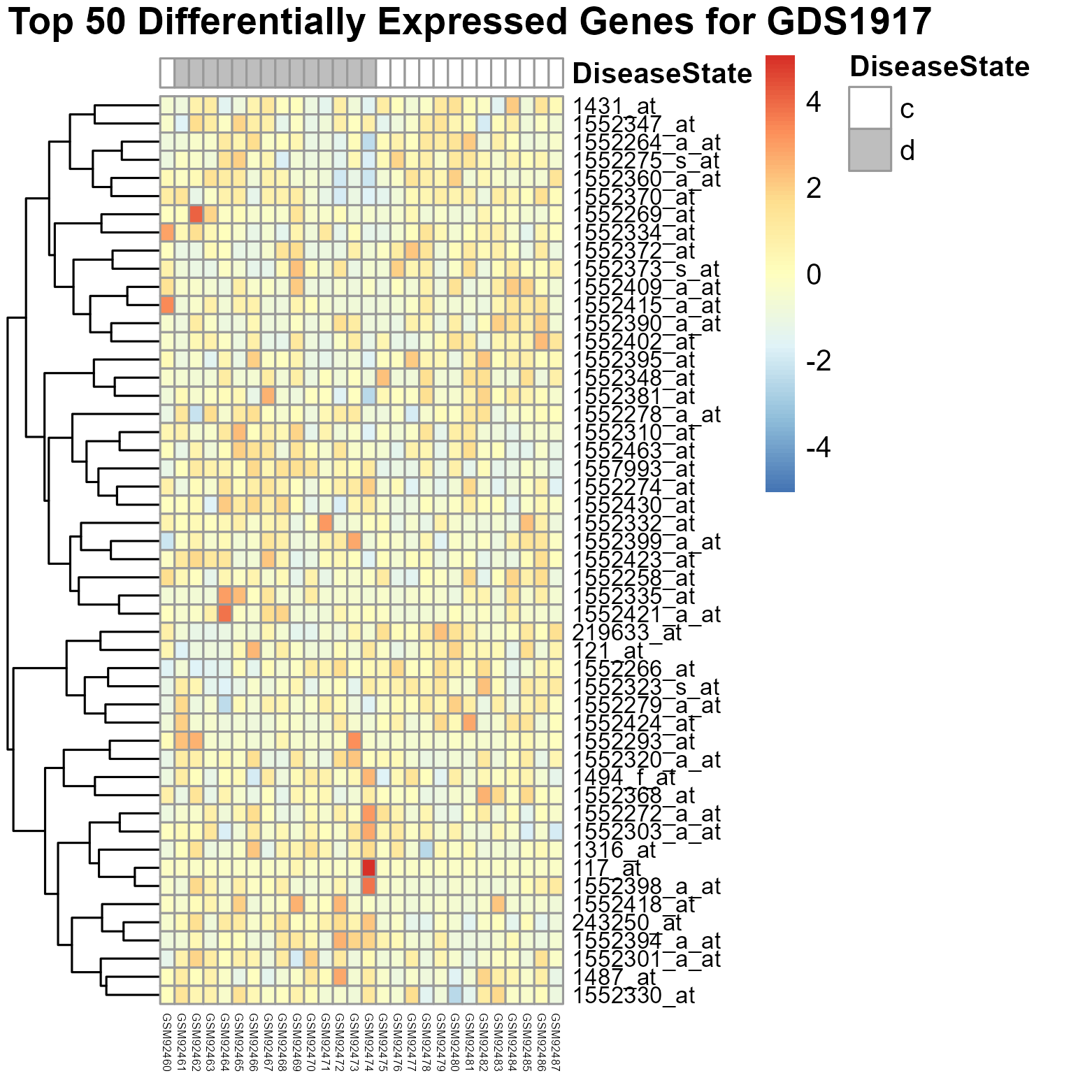
272 KB
{kind=link}
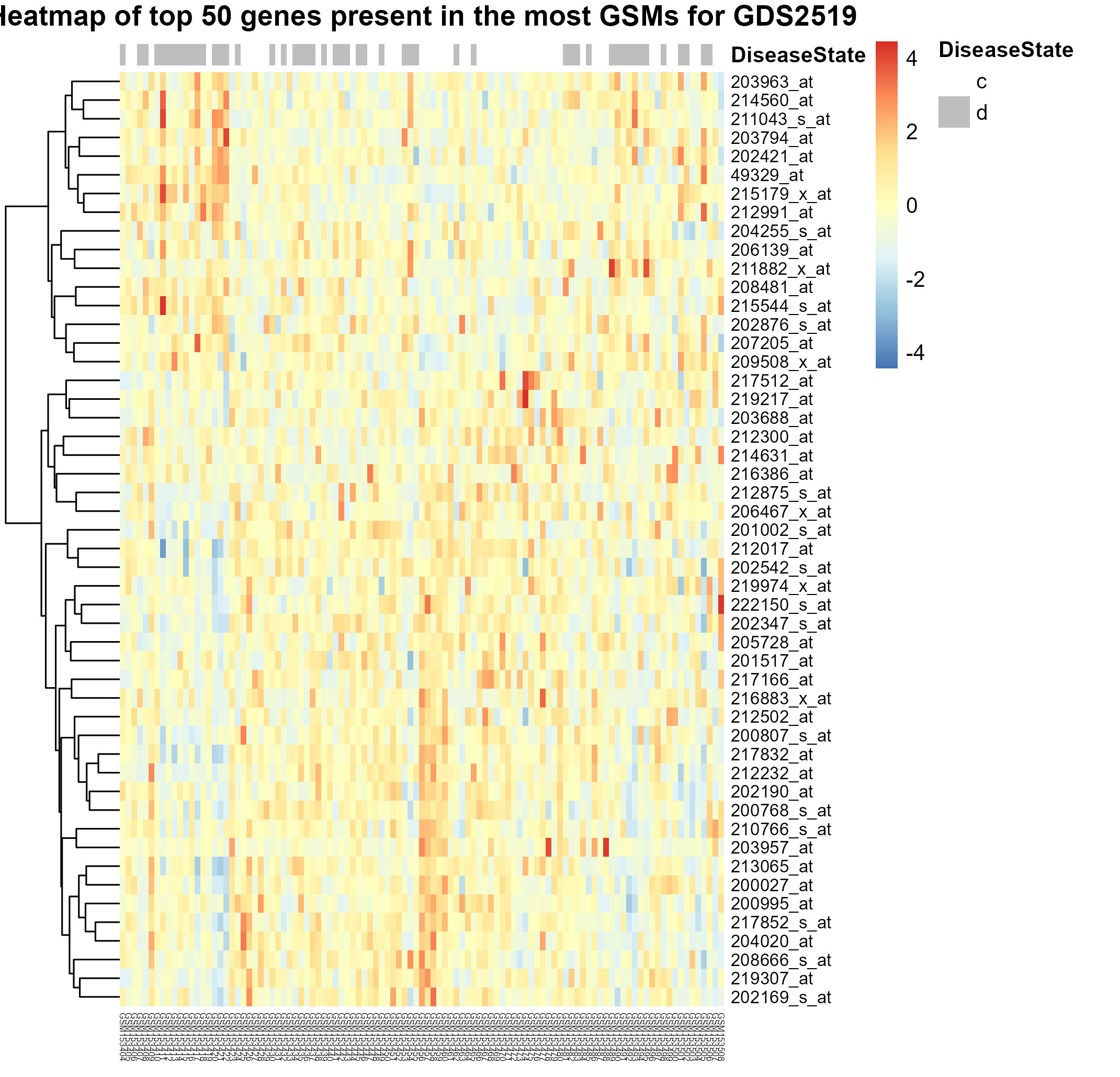
269 KB
{kind=link}
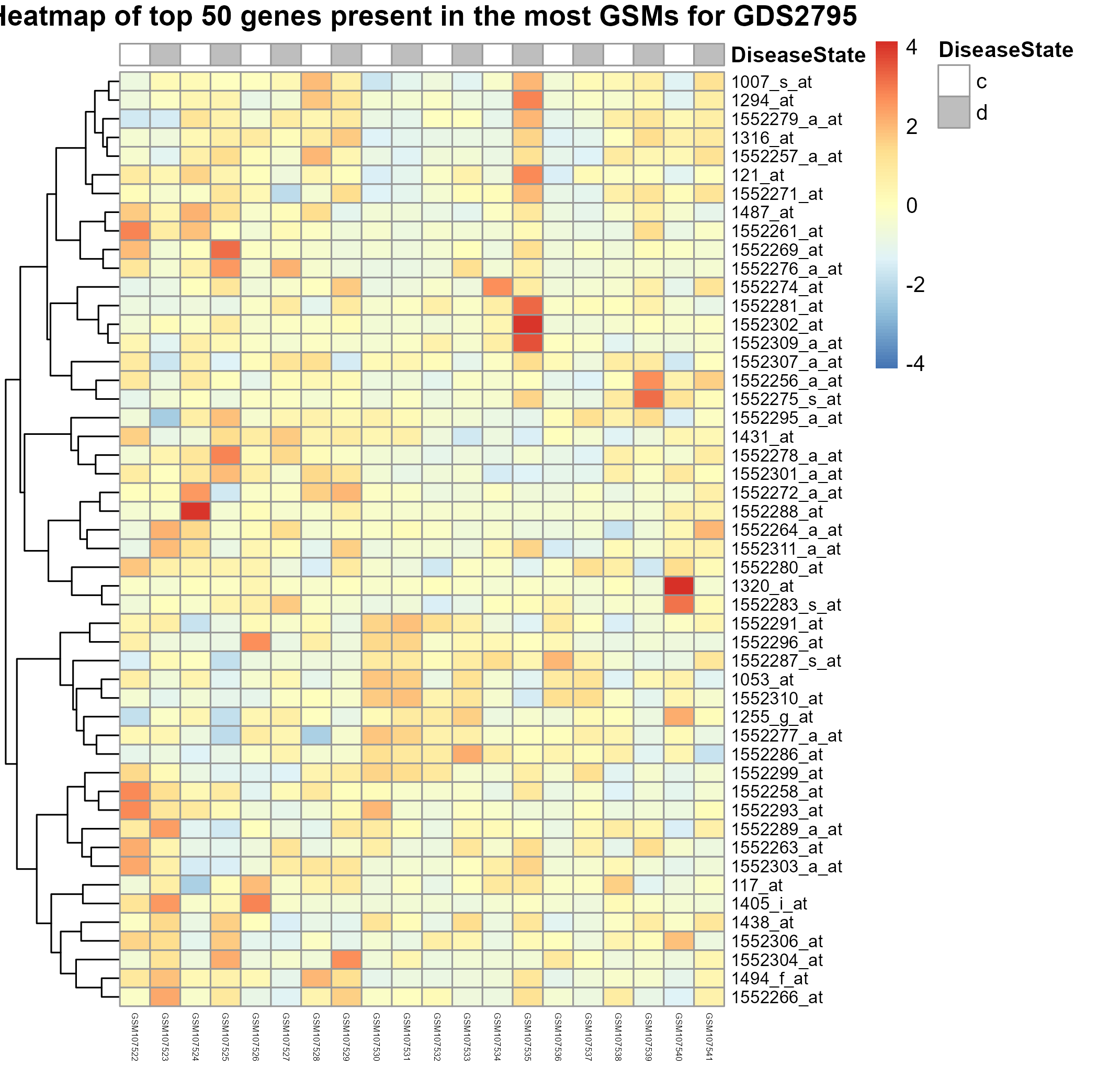
275 KB
{kind=link}
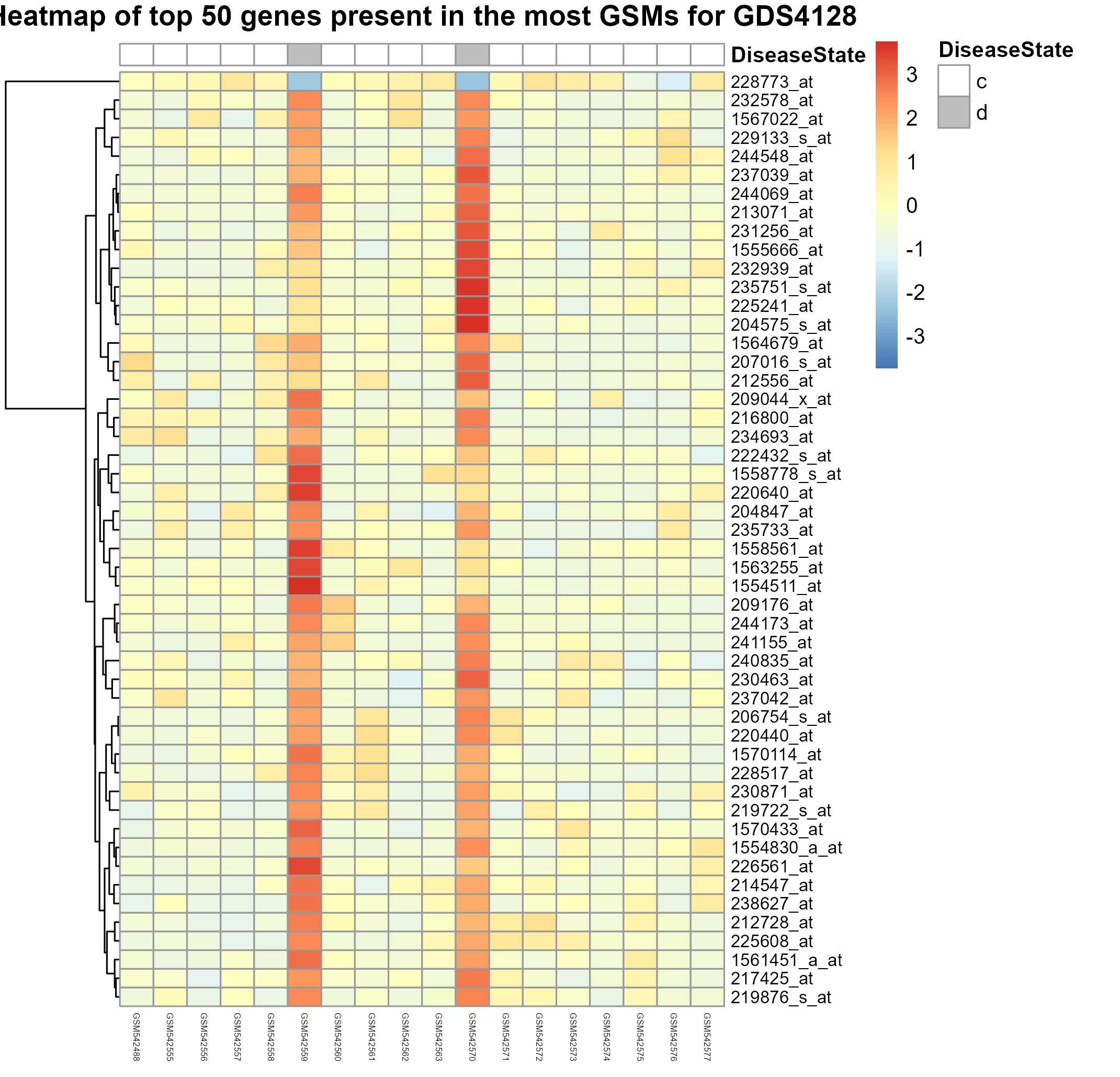
287 KB
{kind=link}
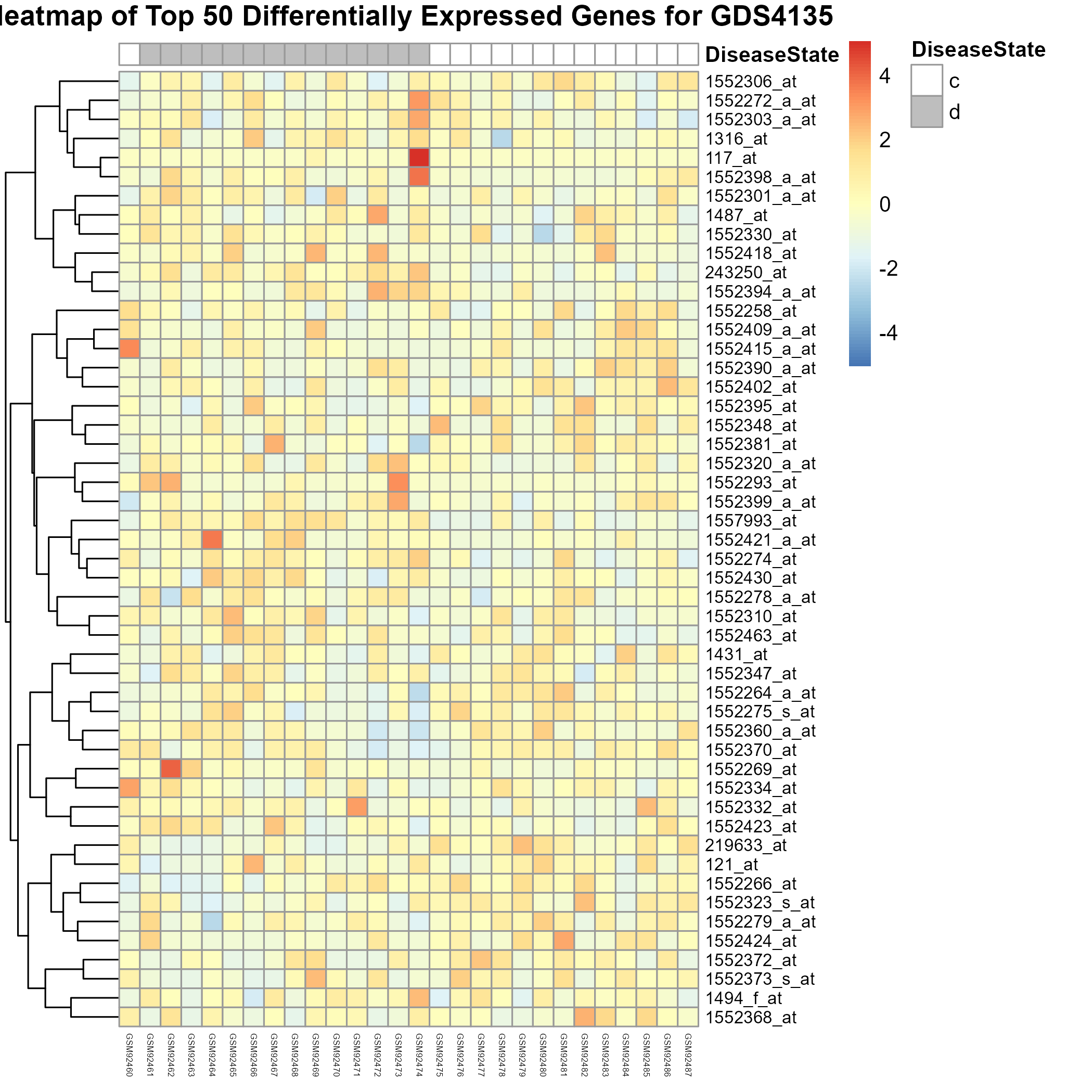
298 KB
{kind=link}
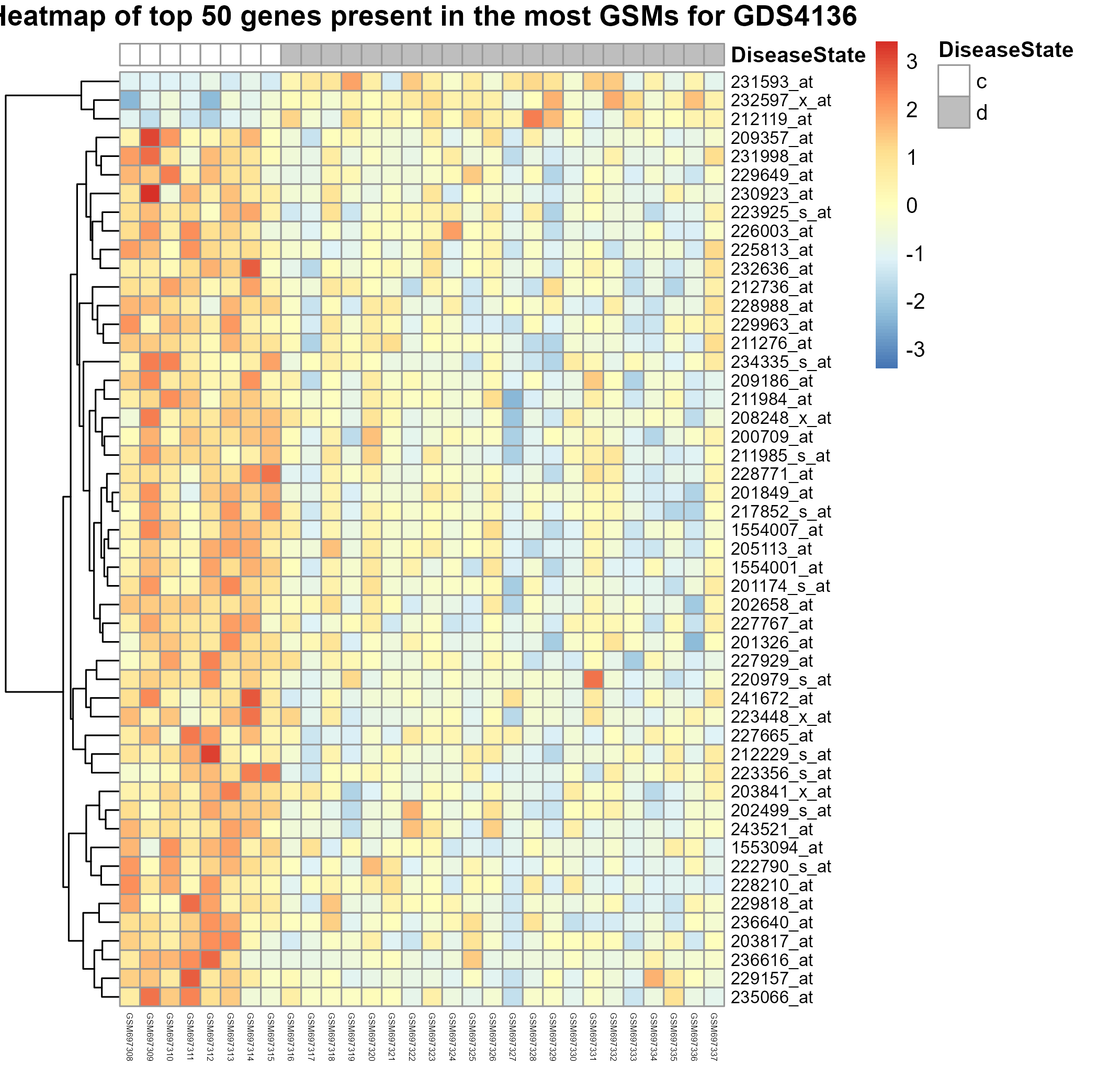
314 KB
{kind=link}
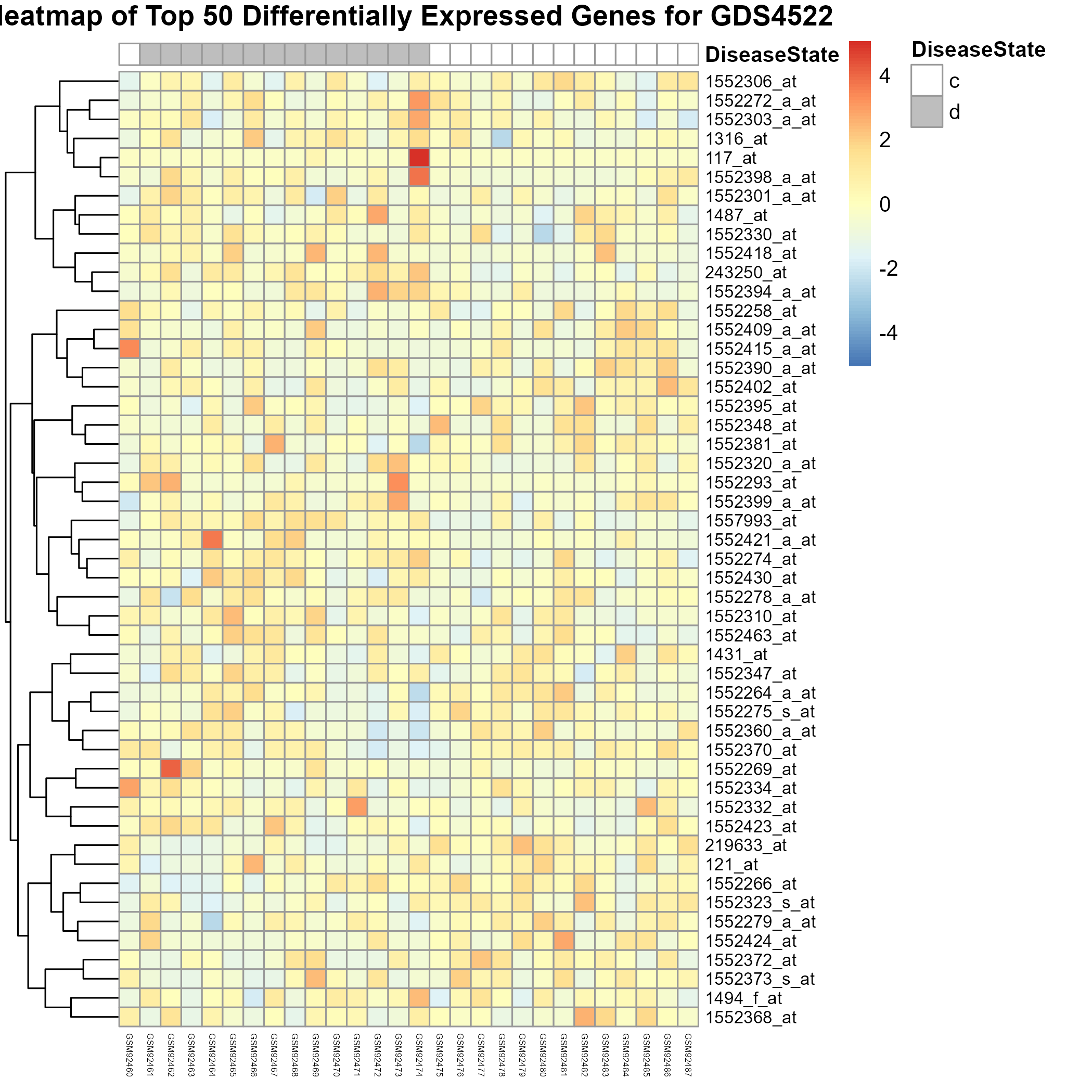
298 KB
{kind=link}
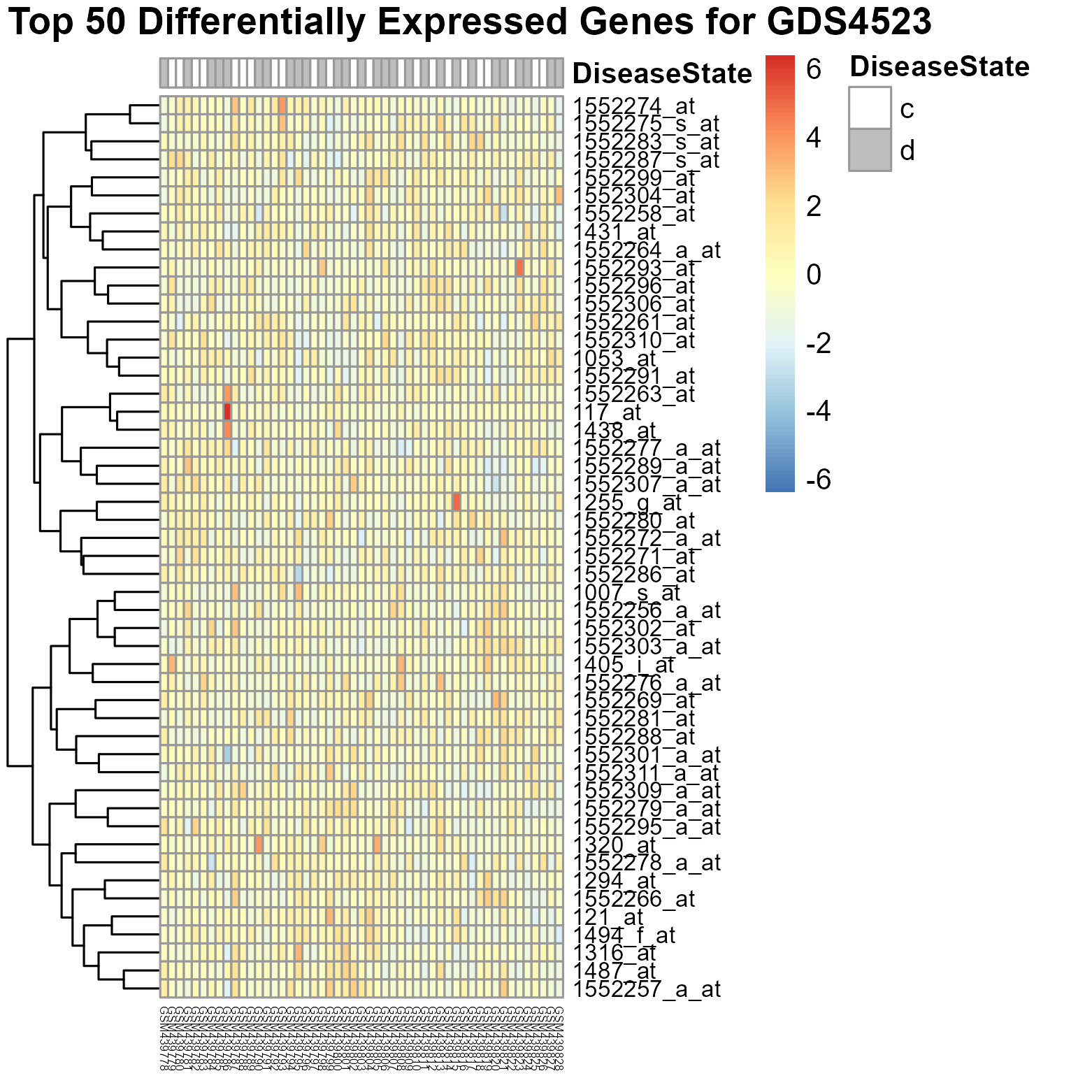
302 KB
{kind=link}
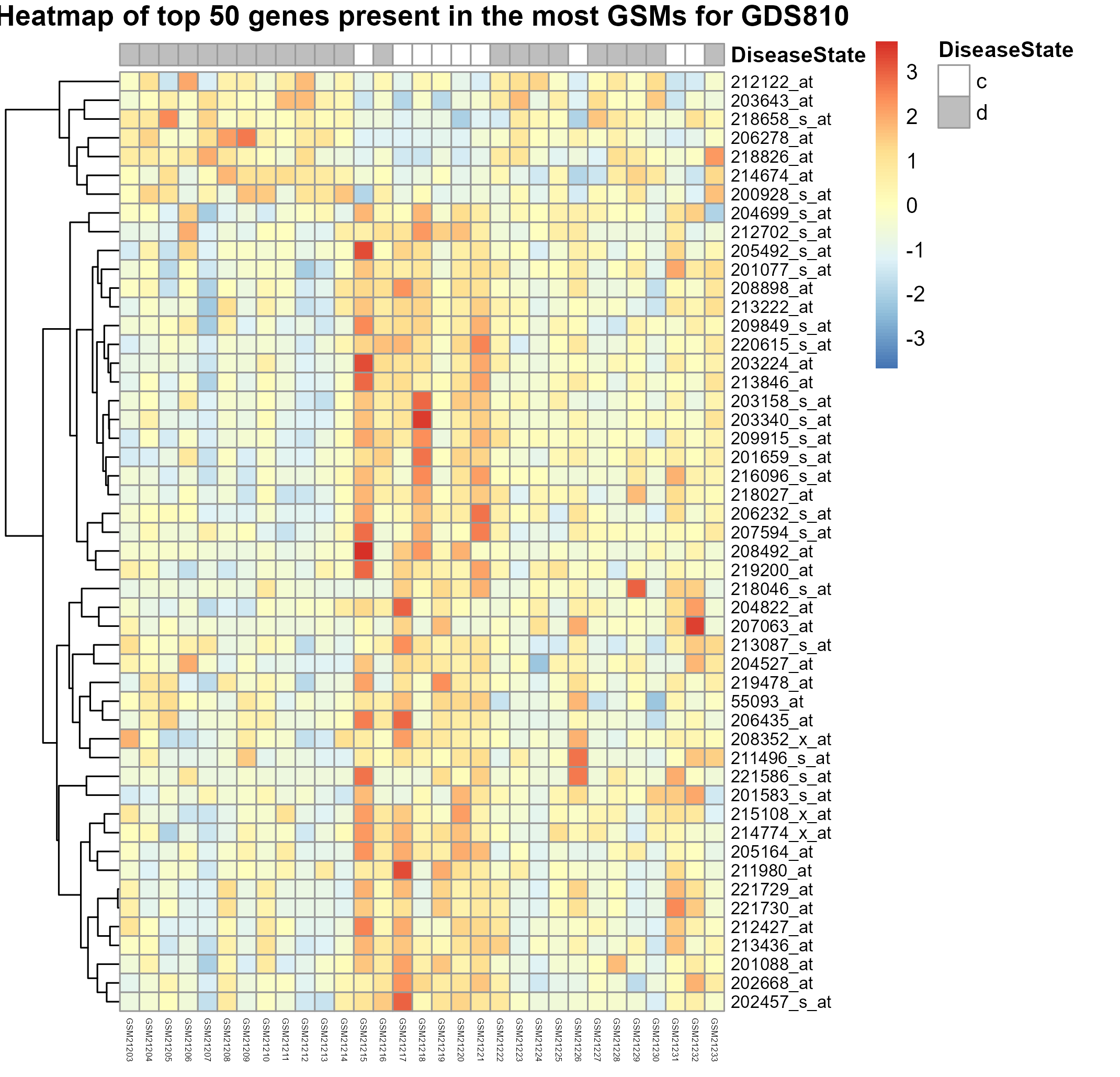
324 KB
{kind=link}
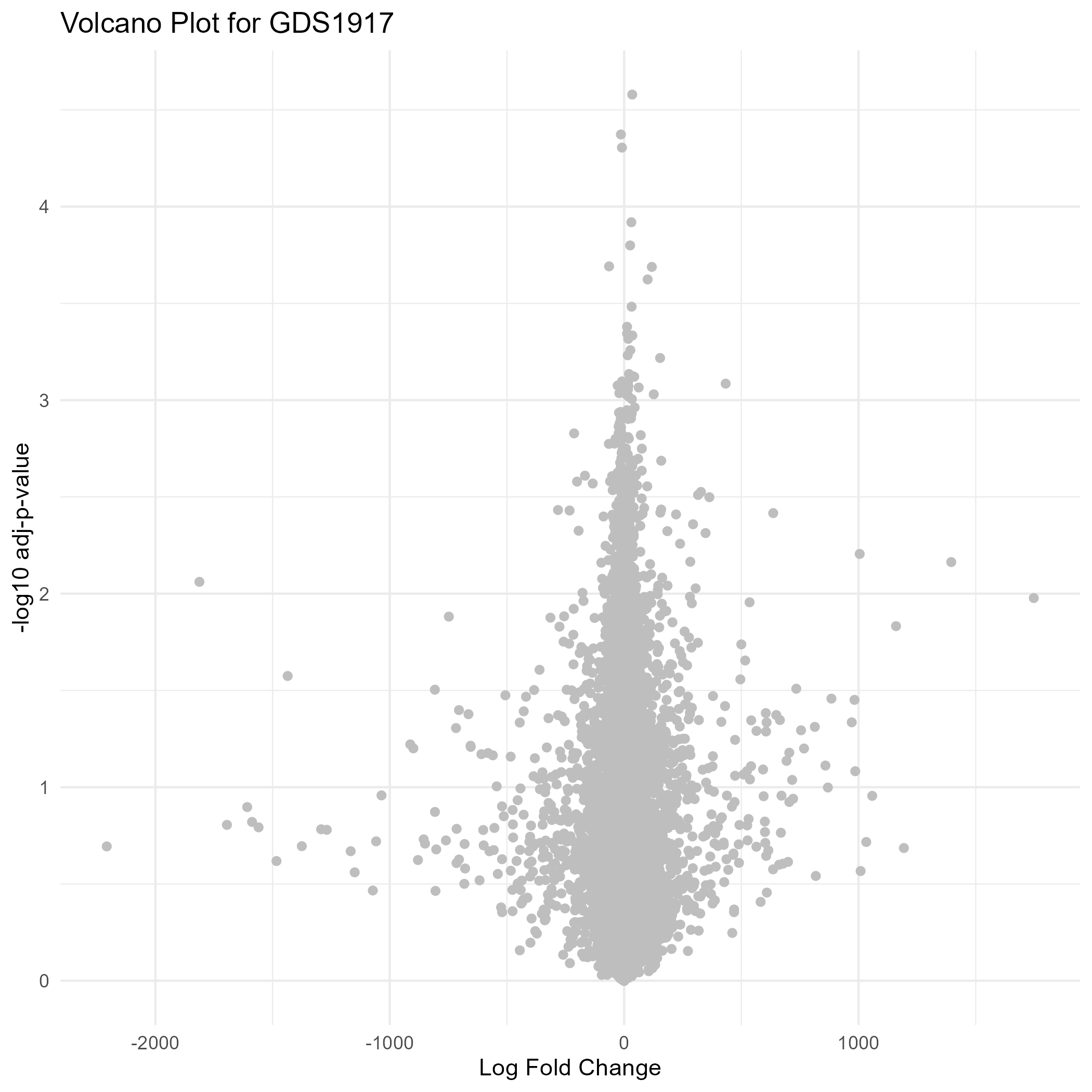
182 KB
{kind=link}
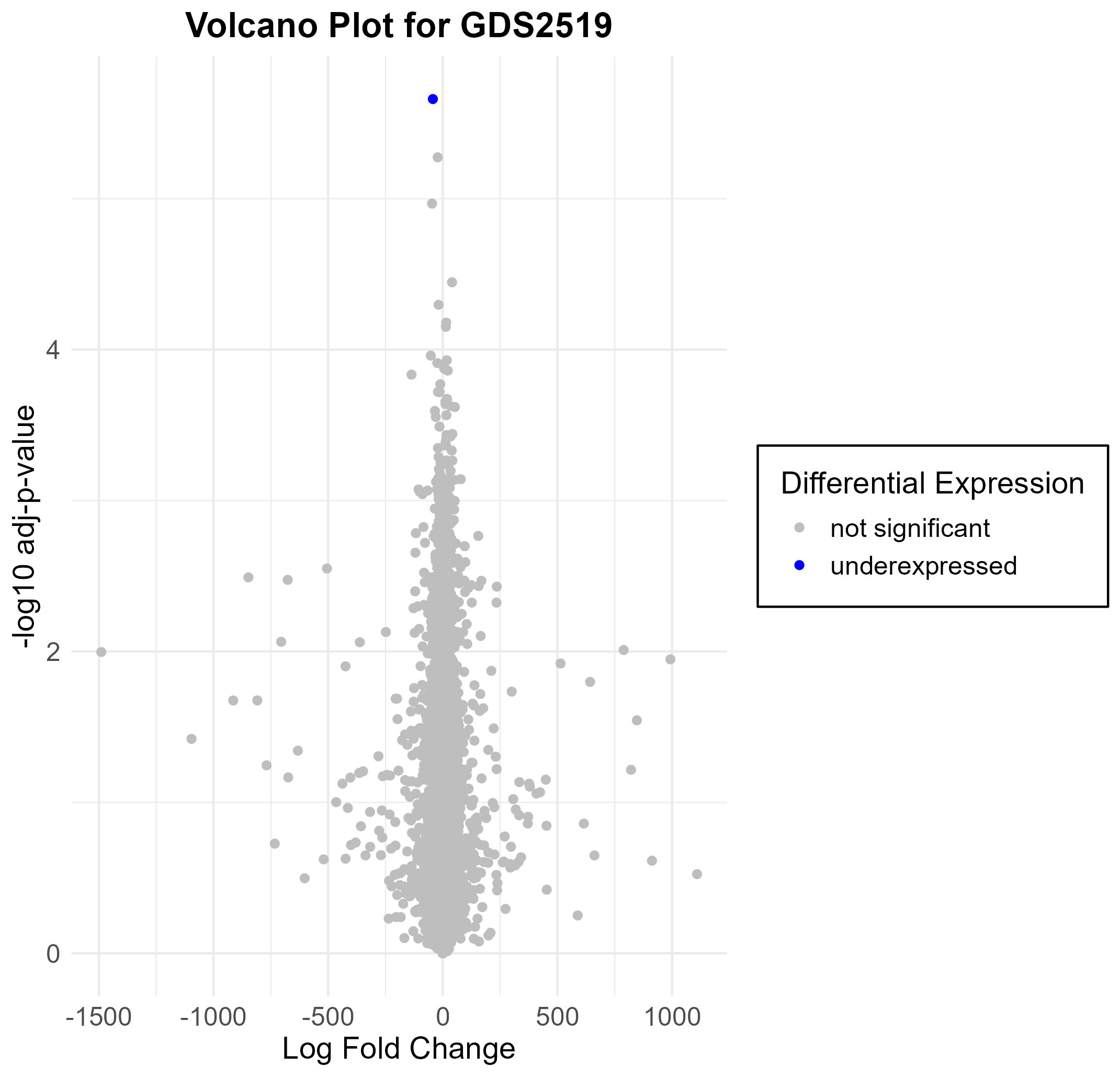
155 KB
{kind=link}
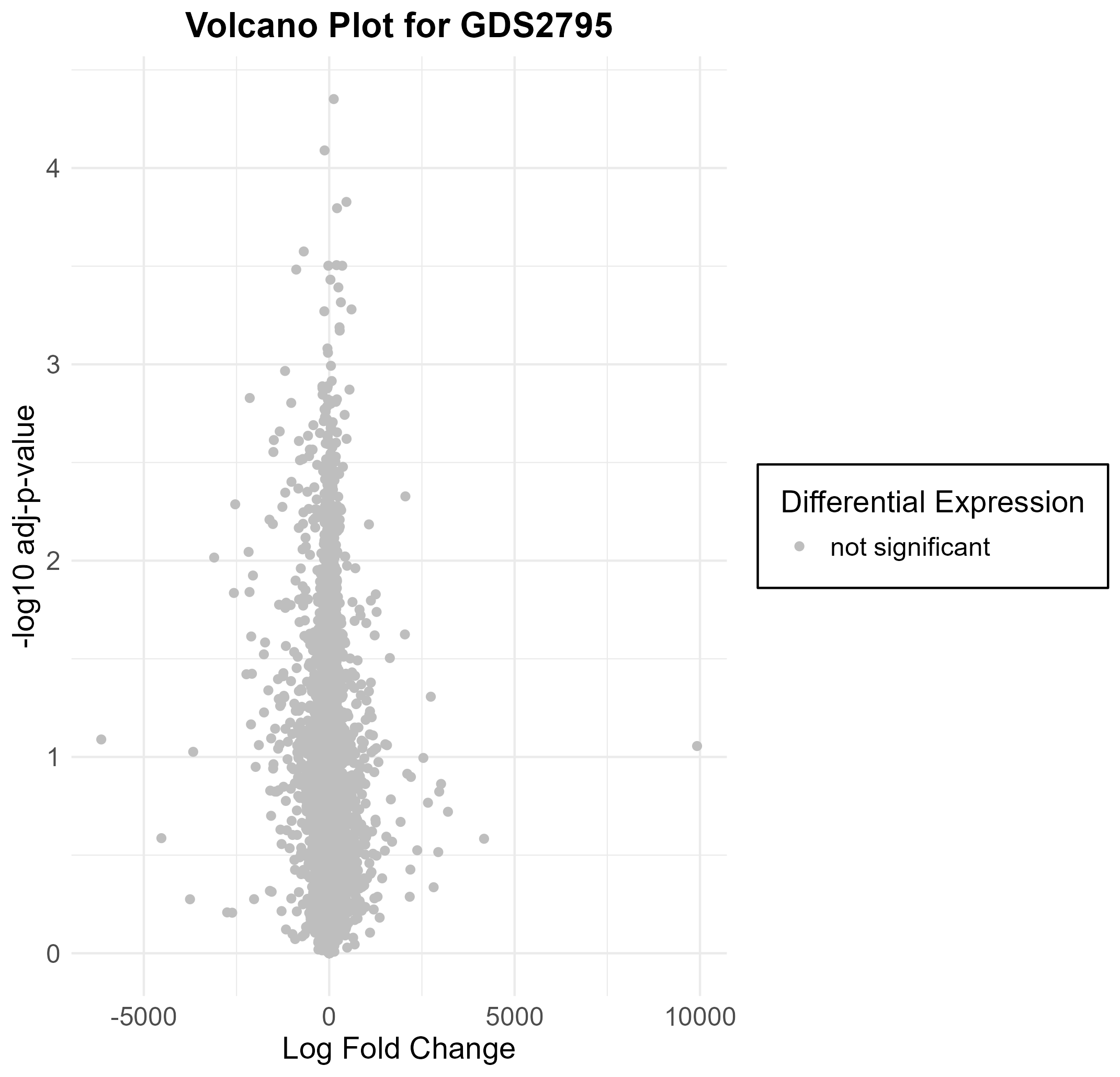
158 KB
{kind=link}
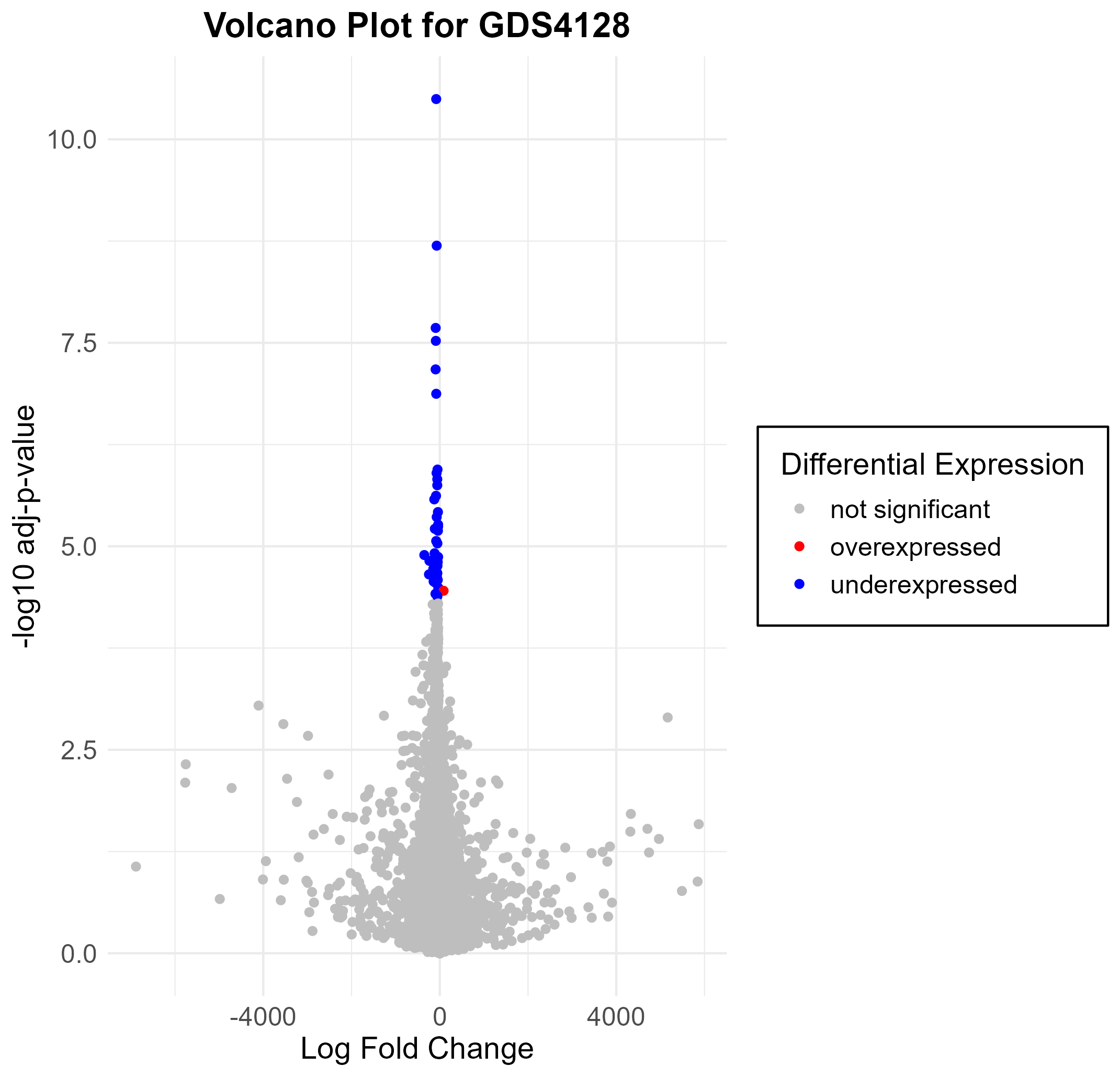
158 KB
{kind=link}
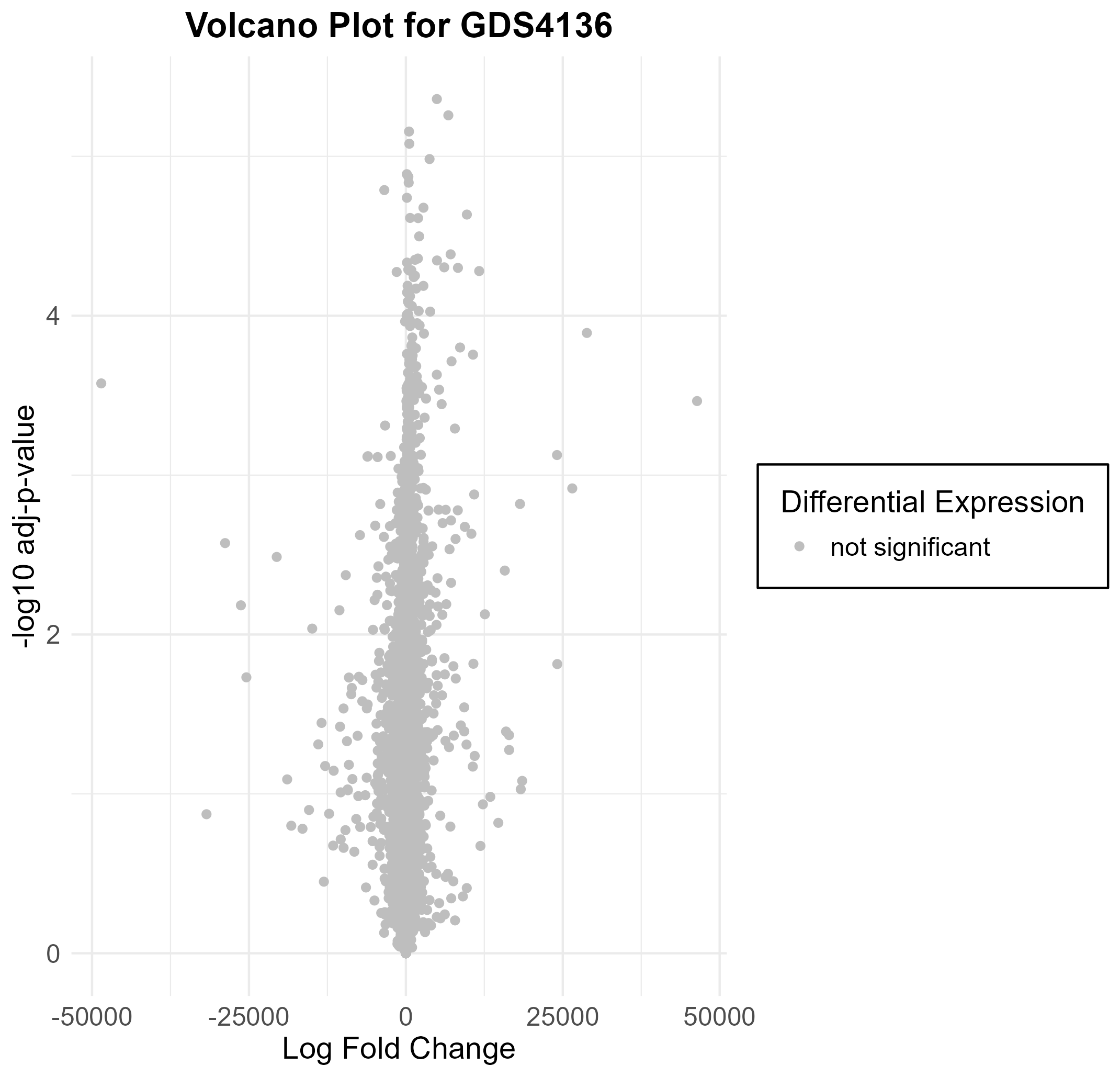
160 KB
{kind=link}
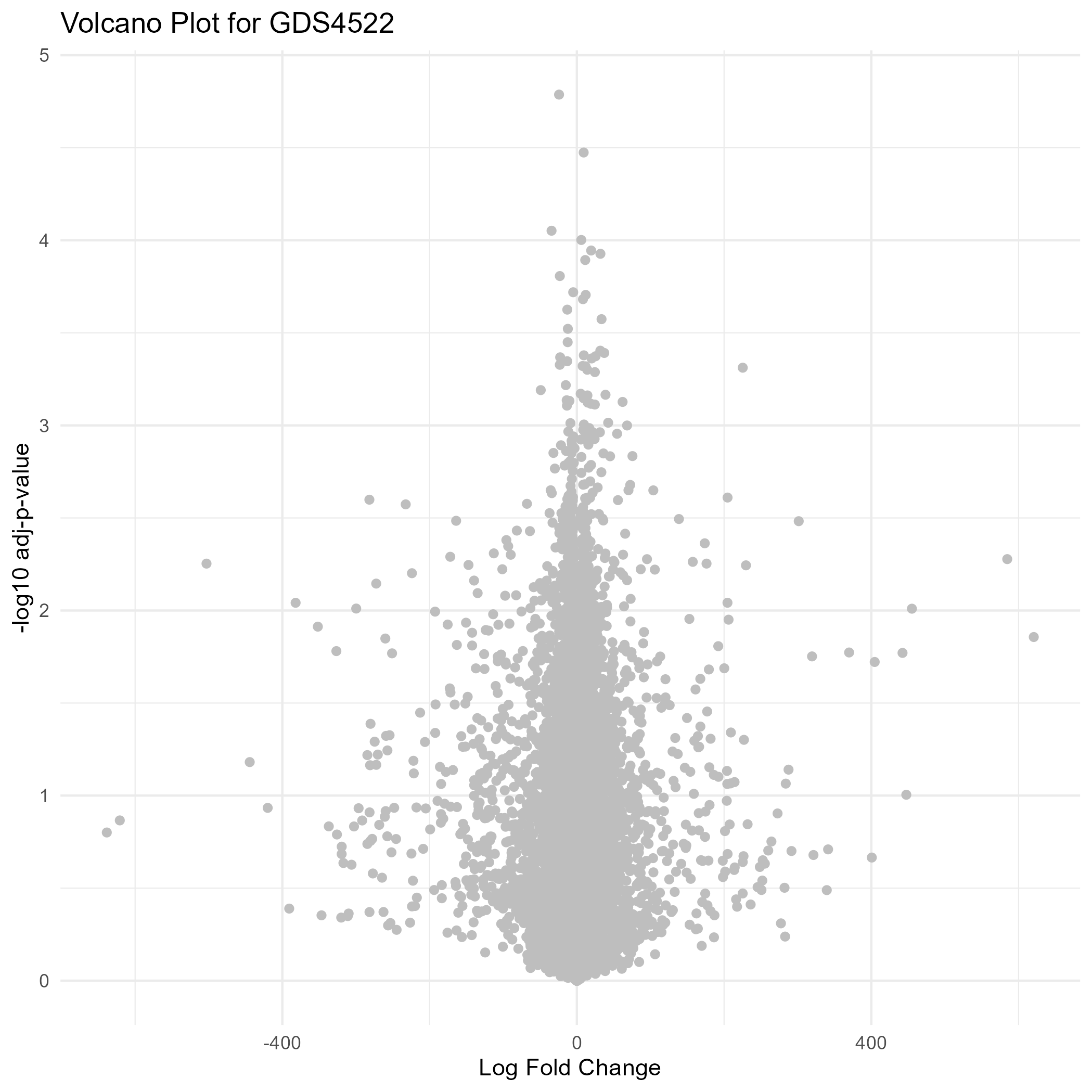
211 KB
{kind=link}
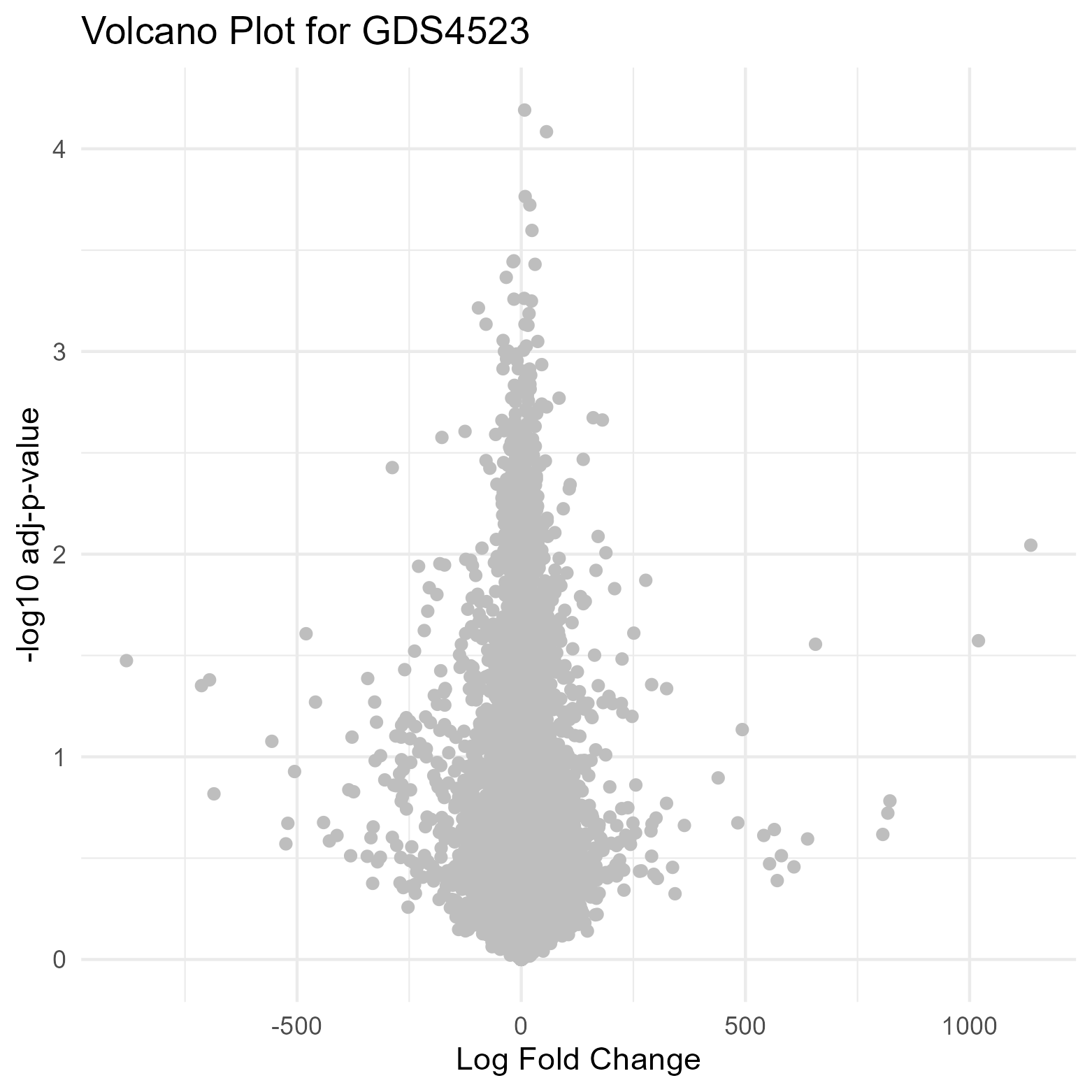
121 KB
{kind=link}
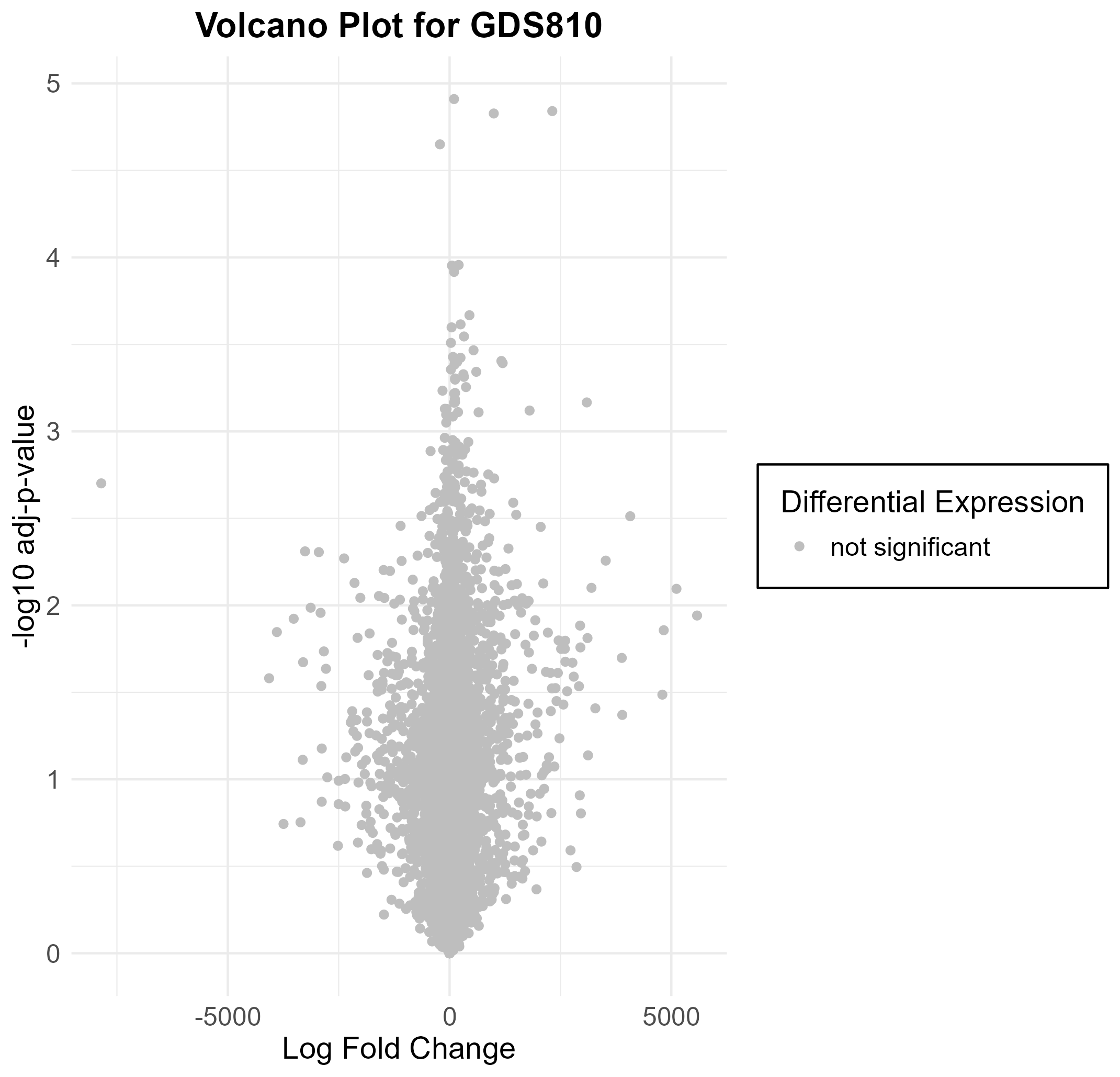
185 KB
This source diff could not be displayed because it is too large. You can view the blob instead.
{kind=link}
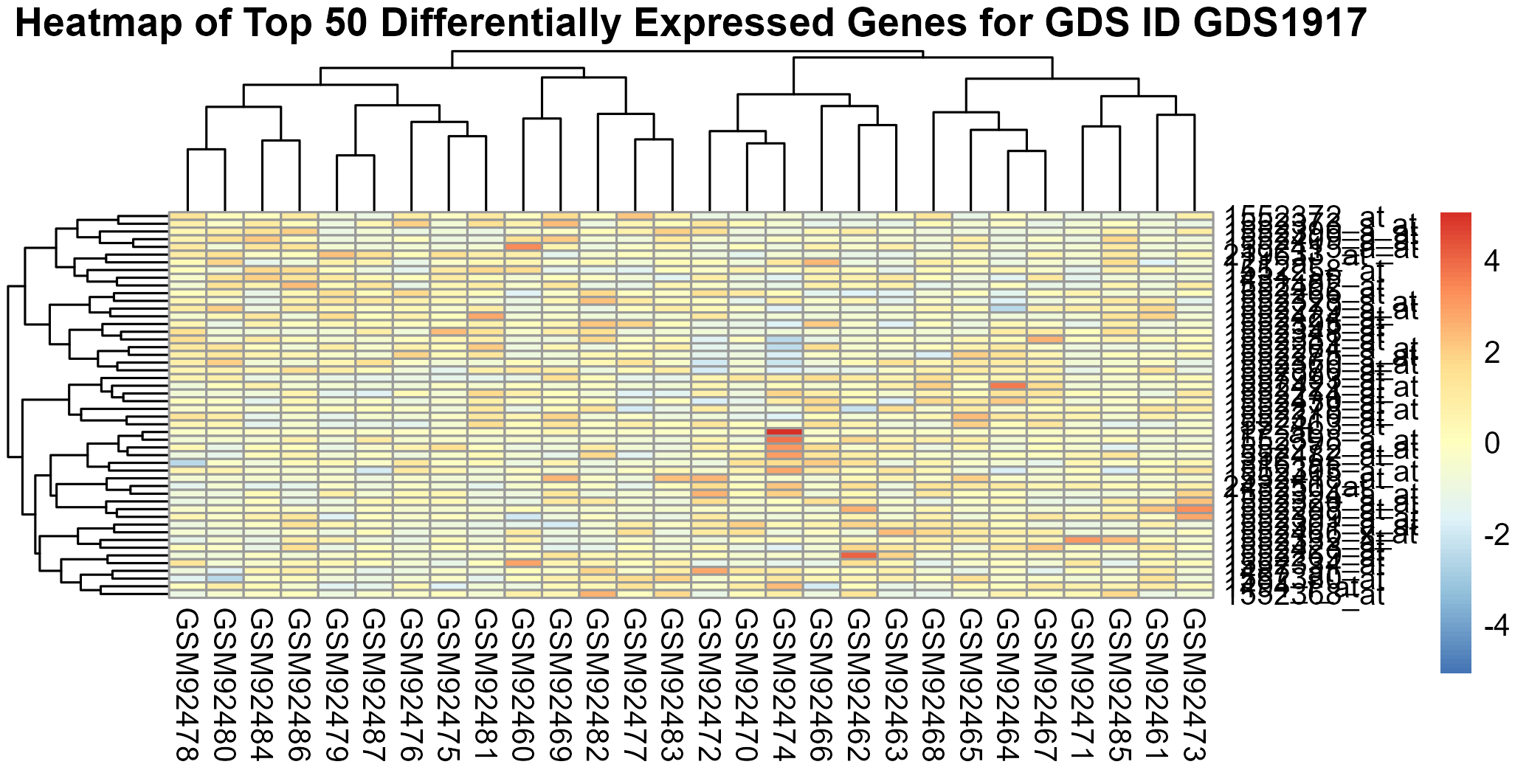
247 KB
{kind=link}
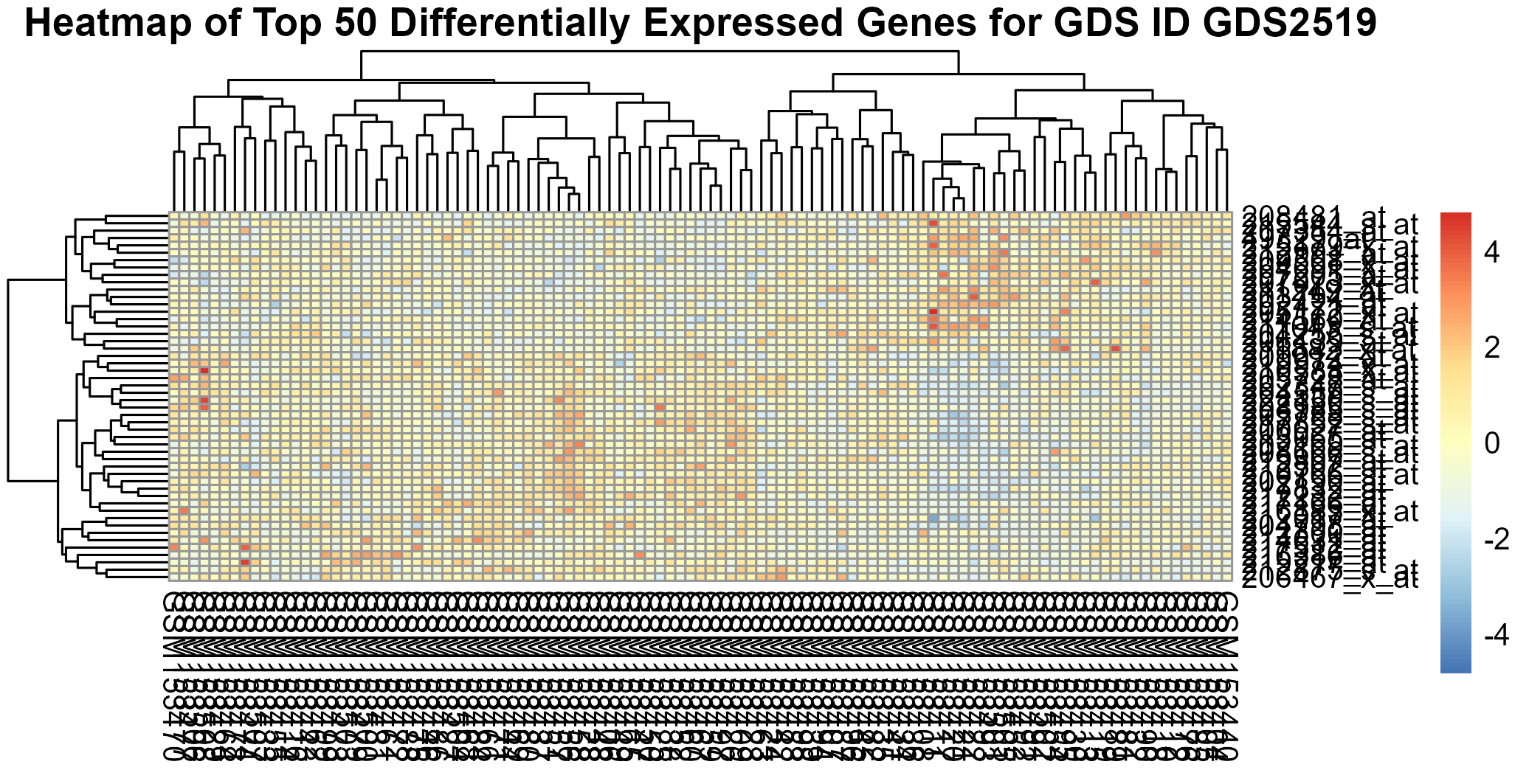
357 KB
{kind=link}
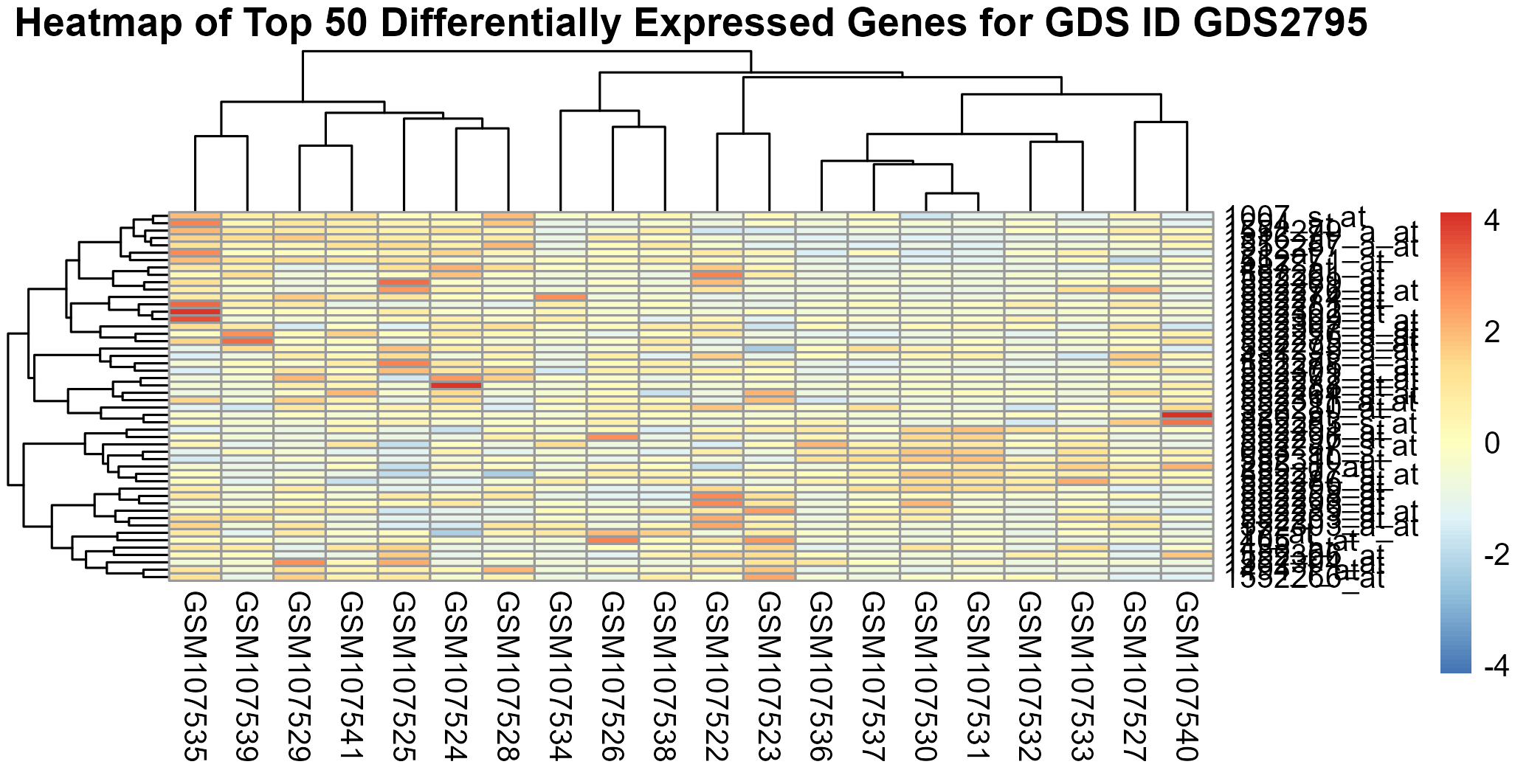
213 KB
{kind=link}
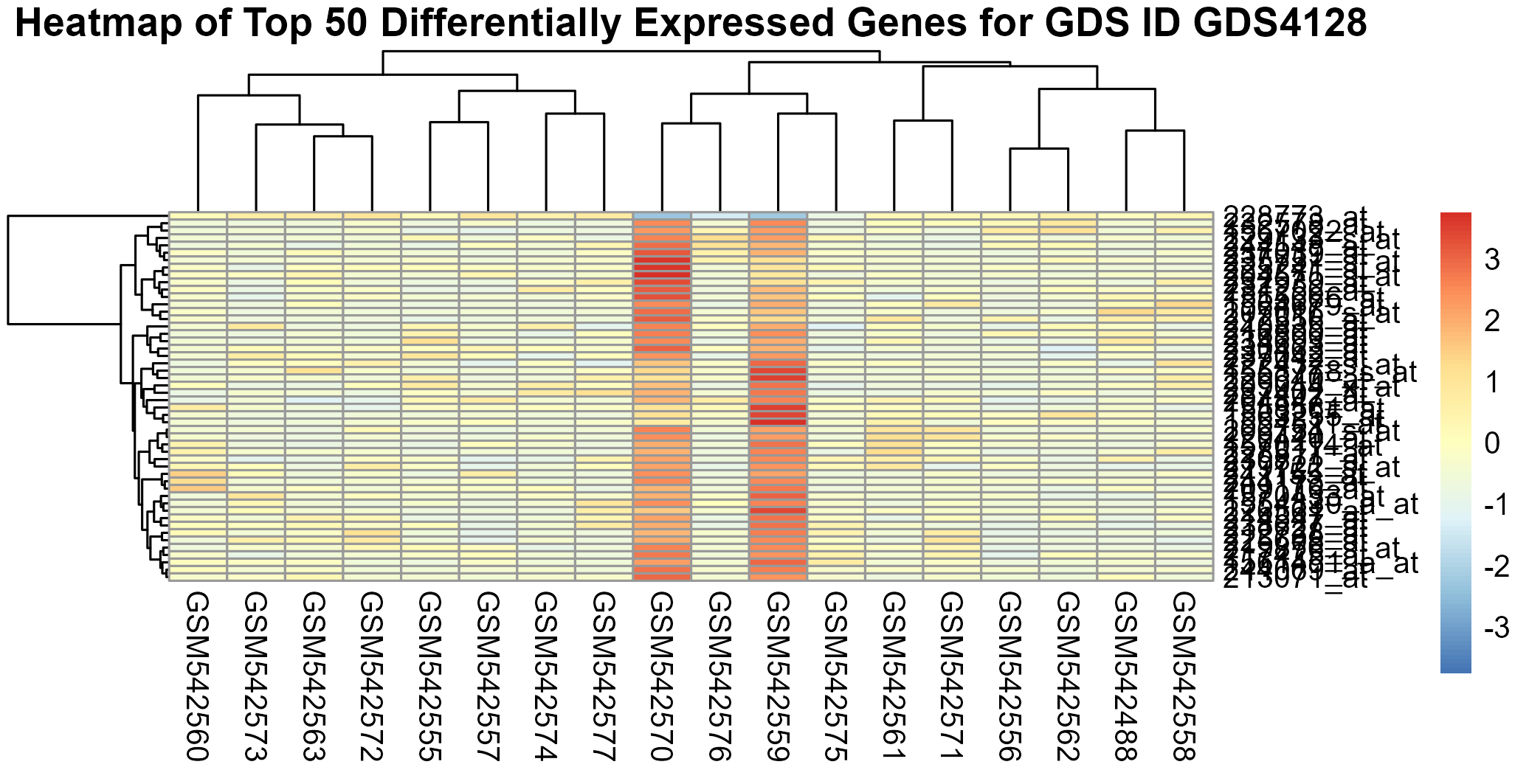
221 KB
{kind=link}
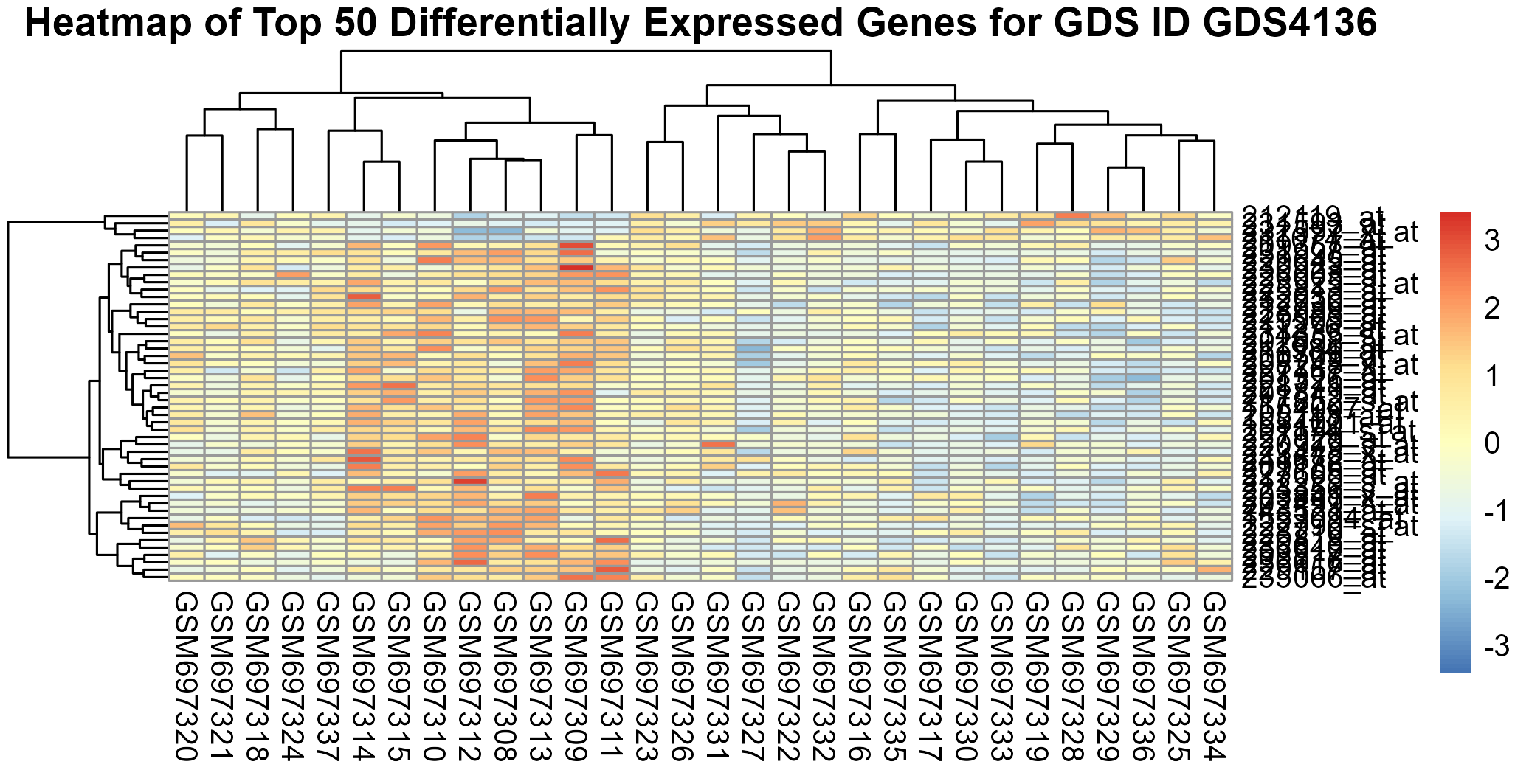
238 KB
{kind=link}
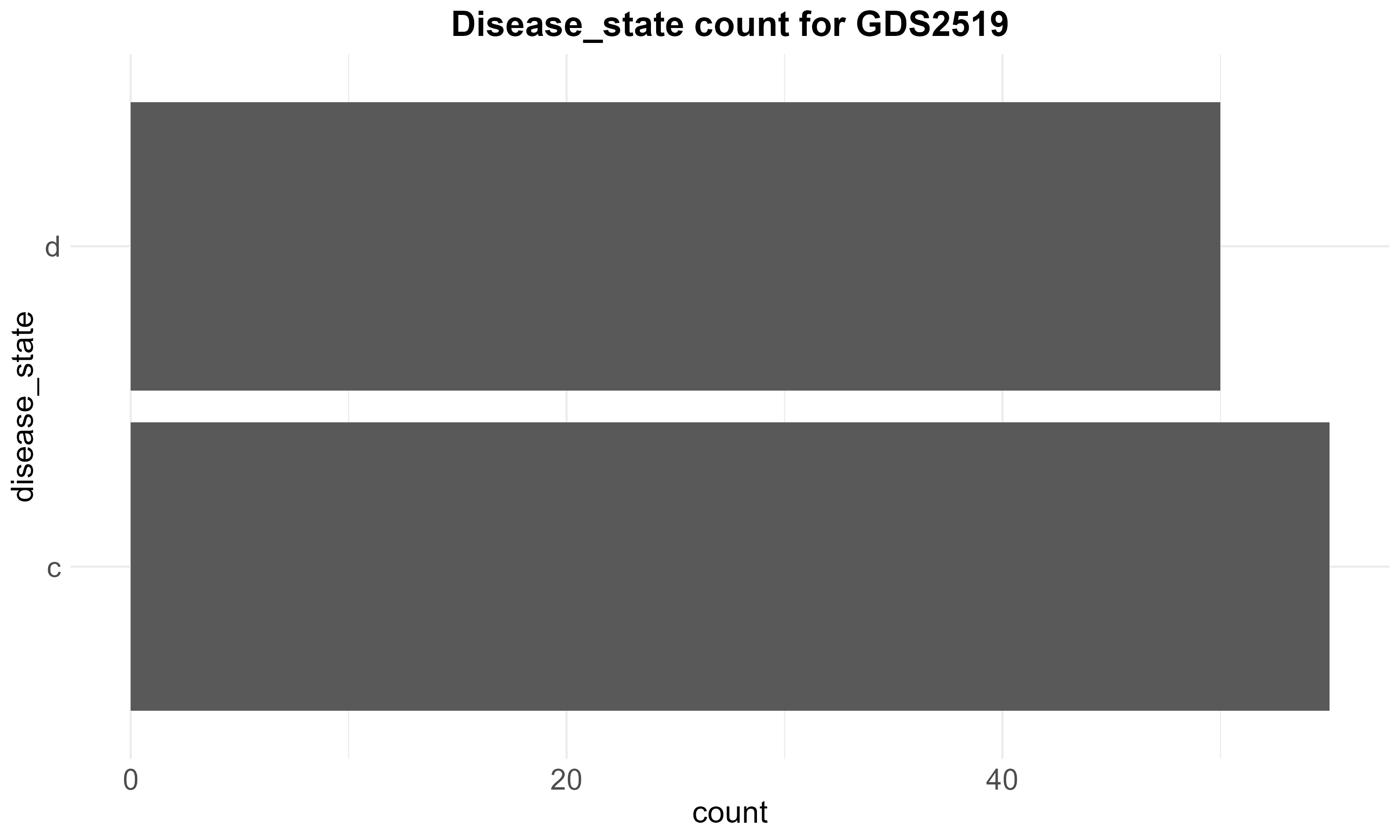
59.7 KB
{kind=link}
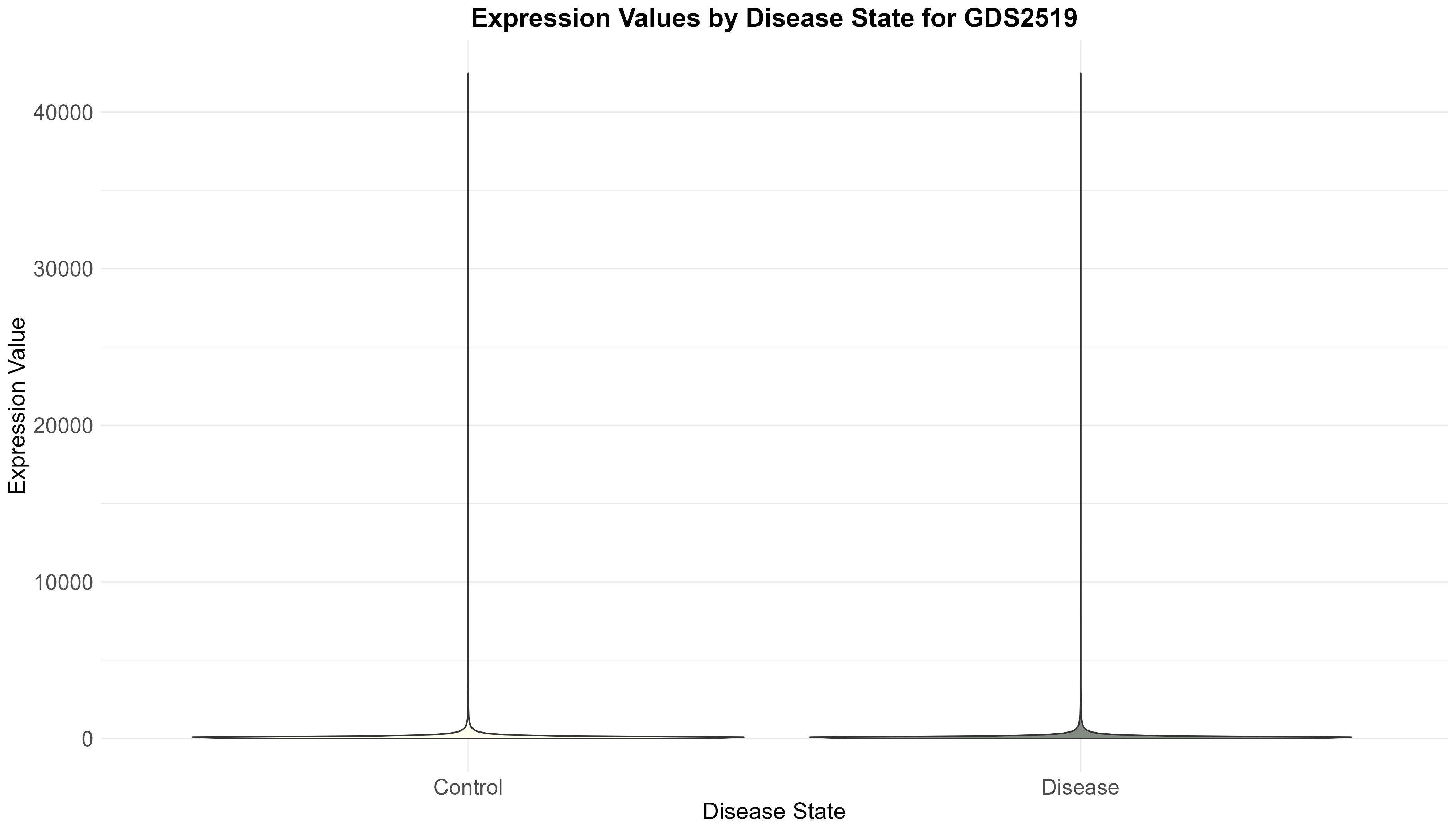
111 KB
{kind=link}
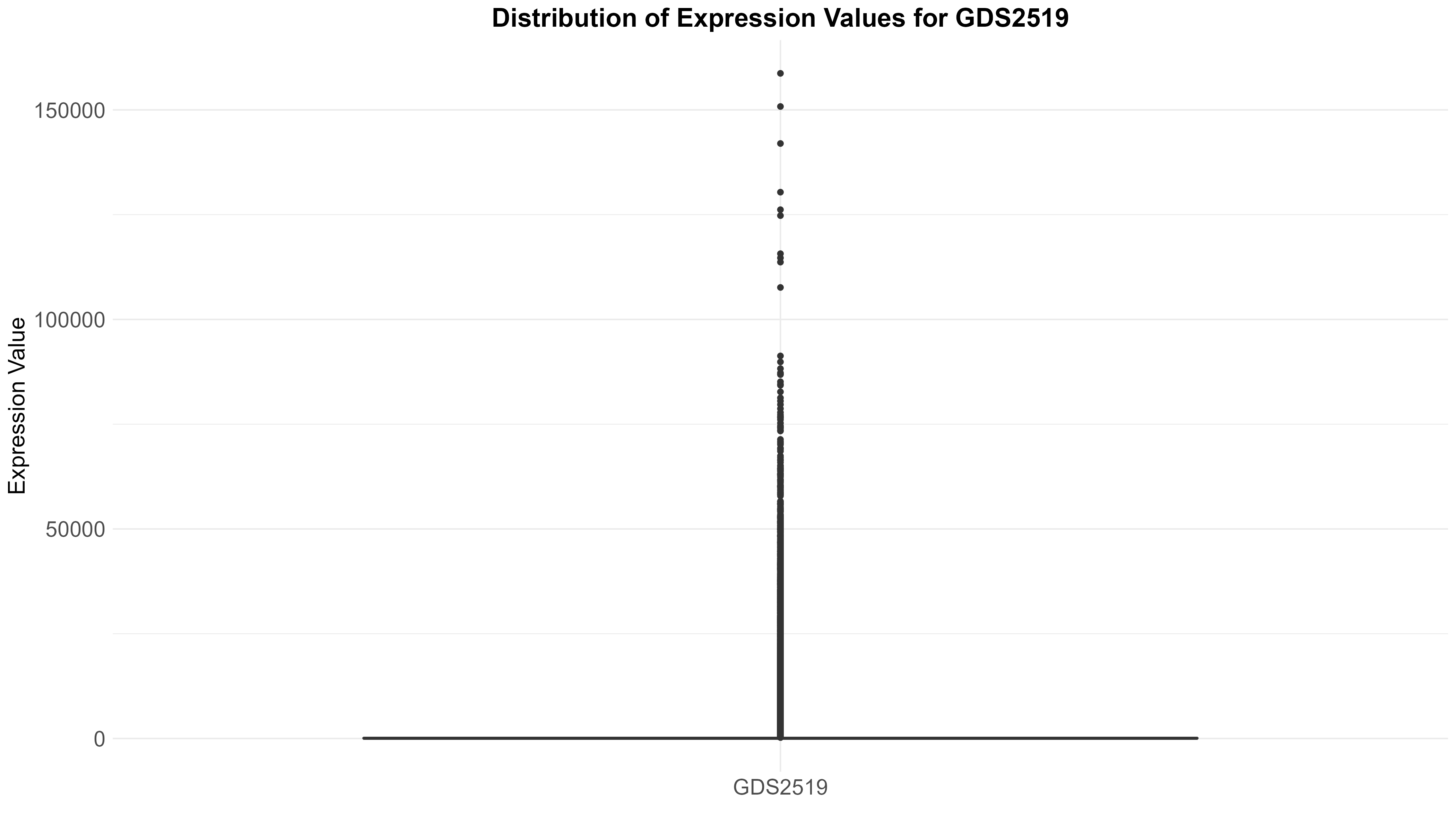
101 KB
{kind=link}
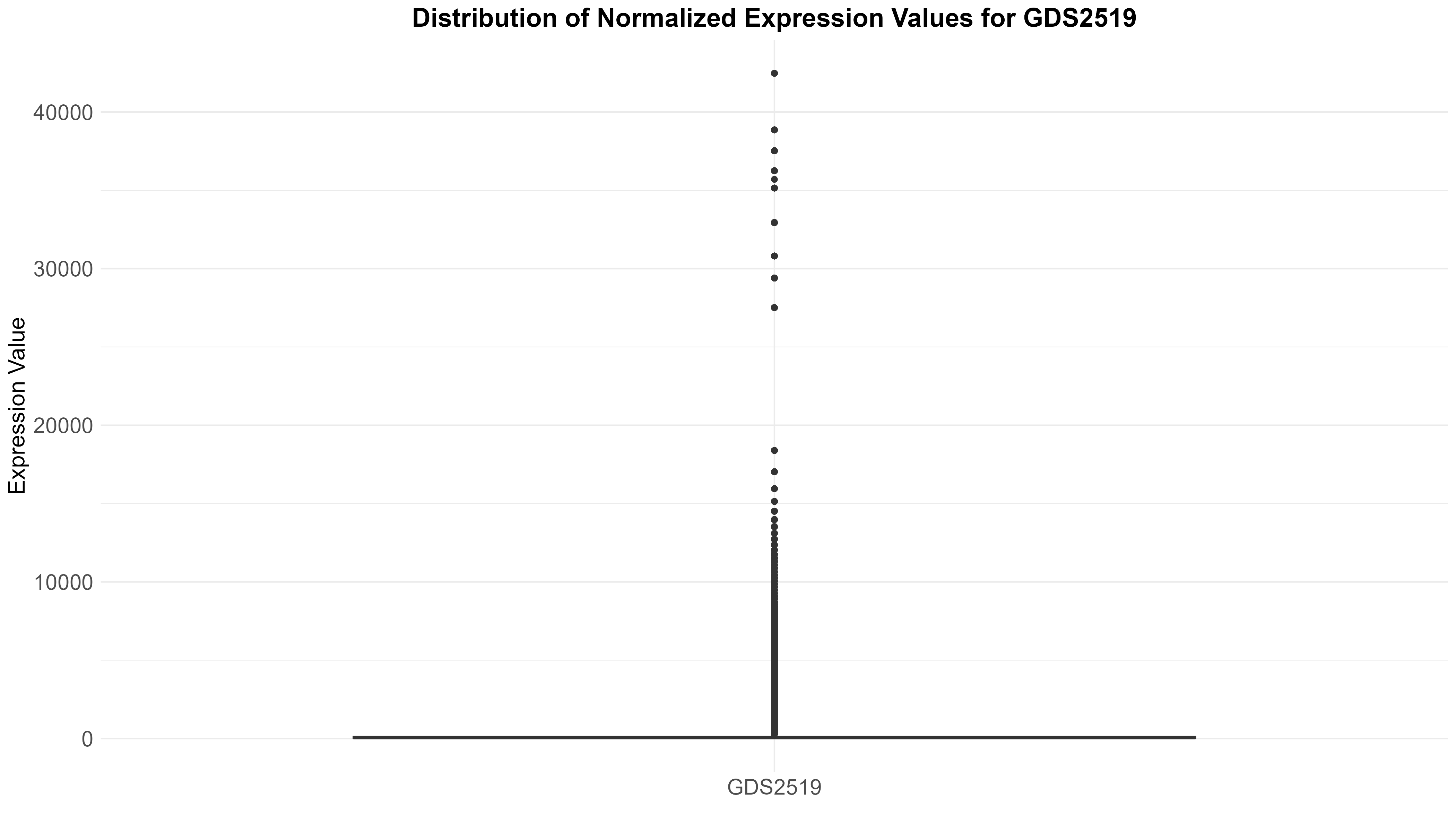
101 KB
{kind=link}
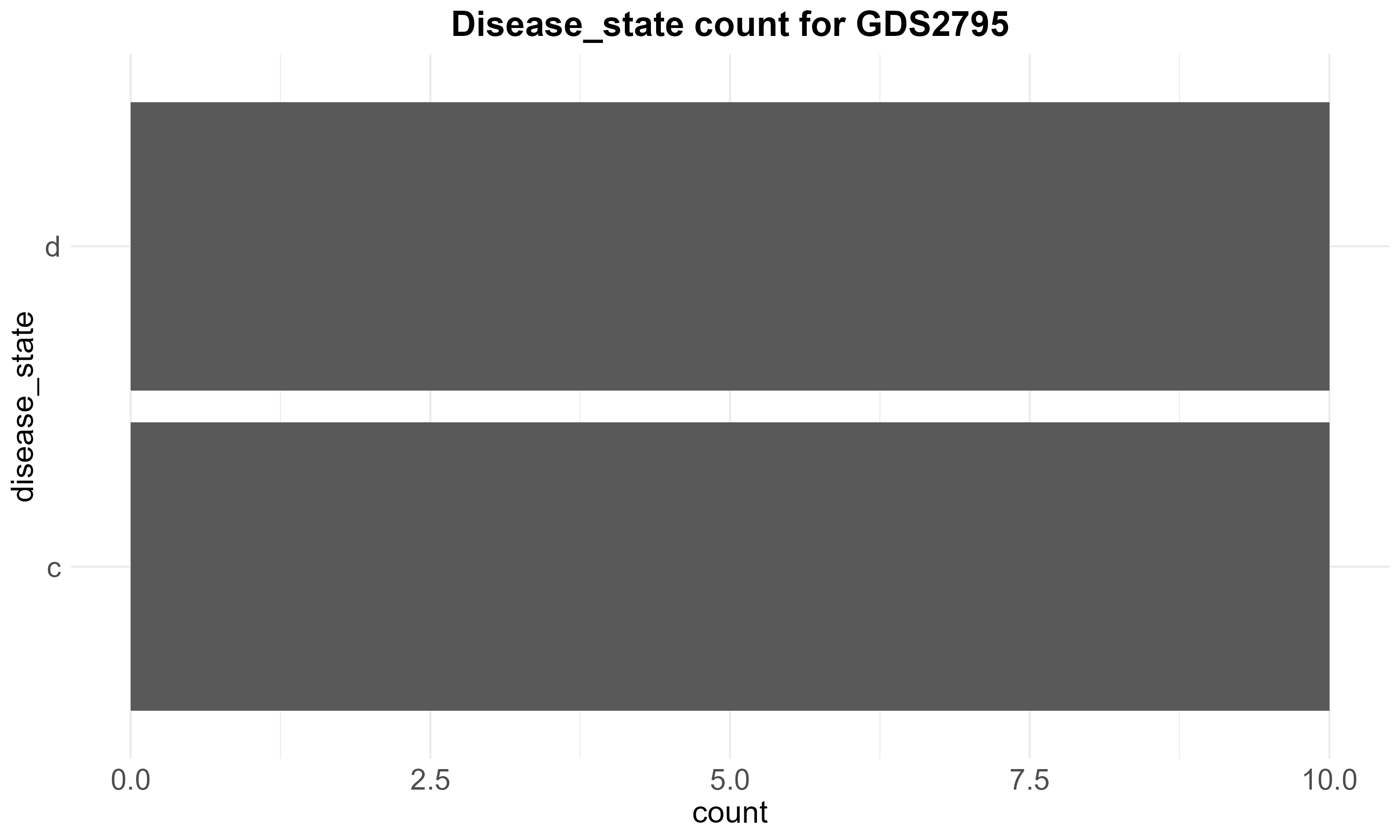
62.3 KB
{kind=link}
114 KB
{kind=link}
105 KB
{kind=link}
105 KB
{kind=link}
59.3 KB
{kind=link}
110 KB
{kind=link}
98 KB
{kind=link}
103 KB
{kind=link}
57.7 KB
{kind=link}
95.3 KB
{kind=link}
101 KB
{kind=link}
95.5 KB
{kind=link}
60.7 KB
{kind=link}
115 KB
{kind=link}
108 KB
{kind=link}
110 KB
{kind=link}
60.2 KB
{kind=link}
123 KB
{kind=link}
98.3 KB
{kind=link}
98.8 KB
{kind=link}
126 KB
{kind=link}
136 KB
{kind=link}
717 KB
{kind=link}
185 KB